
Pathwayとは、解凍系のような細胞の中で起きる連鎖的な化学反応のことです。
サンプル間のトランスクリプトームやプロテオームにおいて有意に変動した遺伝子群が似ていても、Pathwayの観点では似ていないことがあります。
そのため、有意に変動した遺伝子群をPathwayの情報に変換してオミクスの結果を比較することが良く行われています。
本記事ではPathwayの一覧を取得し、さらに各Pathwayが含むタンパクをリストアップする手順を示します。
1. Pathwayのデータベース
有名なPathwayのデータベースといえばKEGGです。
しかし、KEGGは有料であるため基本的には使用できません(※次節)。
そこで、Reactome等の他のPathwayのデータベースが良く利用されます。
本記事では中でもSTRINGからPathwayのデータを取得します。
2. Davidについて
Pathway解析をブラウザから実施可能なWebサイト(David)があります。
DavidではKEGGのデータベースに基づいた解析を提供しているため多くの方が利用しています。
しかし、①APIがないこと、②データが古い可能性がある、という2点の理由からPathway解析には不向きであると考えています。
実験結果を簡単にCheckする程度には良いかもしれませんが、最終的に論文などで報告する際には注意が必要です。
3. STRINGについて
STRINGはタンパク-タンパク相互作用のデータベースという印象が強いですが、そのプログラムの構築に用いたデータセットが公開されており、その中にPathwayのリストが含まれています。
STRINGのPathwayのデータにはReactome等の他のPathwayの情報も含まれているため、STRINGを用いることでより網羅的に解析できると期待できます。
4. STRINGからダウンロード
こちらの9606.protein.enrichment.terms.v12.0.txt.gzという名称のファイルがSTRINGのPathwayに関するデータがまとめられたファイルになります。
上記ファイルをダウンロードして解凍して下さい。
5. STRINGのPathwayデータセットの内容
ファイルをダウンロードし、以下のようにPythonで読み込んで表示します。
import pandas as pd
df_pathways = pd.read_csv('string/9606.protein.enrichment.terms.v12.0.txt', sep='\t')
# preview
df_pathways.head()| #string_protein_id | category | term | description |
| 9606.ENSP00000000233 | Annotated Keywords (UniProt) | KW-0333 | Golgi apparatus |
| 9606.ENSP00000000233 | Annotated Keywords (UniProt) | KW-0342 | GTP-binding |
| 9606.ENSP00000000233 | Annotated Keywords (UniProt) | KW-0449 | Lipoprotein |
| 9606.ENSP00000000233 | Annotated Keywords (UniProt) | KW-0472 | Membrane |
| 9606.ENSP00000000233 | Annotated Keywords (UniProt) | KW-0519 | Myristate |
#string_protein_idにはEmsemblのタンパクIDが記載されており、そのタンパクがtermで定義されるPathwayに含まれることを意味します。
1行目を例にとると、KW-0333というIDのPathwayにはENSP00000000233というIDのタンパクが含まれることになります。
Pathwayの数とタンパクの数は以下のようになっています。
# statistics
pathways = df_pathways.description.drop_duplicates().tolist()
pathways_proteins = list(set([c.split('.')[1] for c in df_pathways['#string_protein_id'].tolist()]))
print(f'n_pathway: {len(pathways)}')
print(f'n_proteins: {len(pathways_proteins)}')n_pathway: 69736
n_proteins: 19647categoryの列には、該当の行がどのデータベース由来の情報であるかを示します。UniProtやReactomeなどが例です。一覧は以下となります。
pathways_categories = df_pathways.category.drop_duplicates().tolist()
print(f'n_database: {len(pathways_categories)}')n_database: 14# database list pathways_categories
['Annotated Keywords (UniProt)',
'Biological Process (Gene Ontology)',
'Cellular Component (Gene Ontology)',
'Local Network Cluster (STRING)',
'Molecular Function (Gene Ontology)',
'Protein Domains and Features (InterPro)',
'Reactome Pathways',
'Tissue expression (TISSUES)',
'WikiPathways',
'Disease-gene associations (DISEASES)',
'Human Phenotype (Monarch)',
'Protein Domains (Pfam)',
'Subcellular localization (COMPARTMENTS)',
'Protein Domains (SMART)']termは由来のデータベースにおけるPathwayのIDです。
descriptionはPathwayに関する説明です。
6. Pathway ID → Symbol の辞書を作成
上記のテーブルを利用し、termをkeyにし、termに含まれるタンパクのシンボルのリストをvalueとする辞書を作成します。
まずは上記テーブルの#string_protein_idから、以下のようにユニークなemsembl IDのリストを作成して書き出します。
f = open('id_conversion/pathway_protein_ensemblP.csv', 'w')
f.writelines(['ensemblP_id\n'] + [str(i) + '\n' for i in pathways_proteins])
f.close()次にEmsembl IDからARF5のような遺伝子名(シンボル表記という)に変換します。
変換にはこちらの記事のように予めRのbiomaRtから作成したした変換テーブル(ensemblP_to_symbols.csv)を作成しておき、以下のように実施します。ついでにpickleで保存もしておきます。
import pickle
# conversion
df_eid_sym = pd.read_csv('id_conversion/ensemblP_to_symbols.csv', index_col=0)
eid_sym = df_eid_sym['uniprot_gn_symbol'].to_dict()
# save
with open('temp/ensemblProtein ID_to_symbol.pkl', 'wb') as tf:
pickle.dump(eid_sym, tf)最後に以下のようにして、Pathway ID → シンボル の辞書を作成して保存します。
# dict: pathway term --> symbols
def to_eid(spid):
return spid.split(".")[1]
df_pathways['symbol'] = df_pathways['#string_protein_id'].apply(
lambda x: eid_sym[to_eid(x)] if to_eid(x) in eid_sym.keys() else np.nan
)
ptm_syms = makeList_groupby(df_pathways, 'term', 'symbol', unique=True)
# save
TEMP_DIR = 'temp'
with open(f'{TEMP_DIR}/pathwayTerm_to_symbols.pkl', 'wb') as tf:
pickle.dump(ptm_syms, tf)なお、pickleで保存した辞書は以下のようにしてロードします。
# load
with open(f'{TEMP_DIR}/pathwayTerm_to_symbols.pkl', 'rb') as tf:
ptm_syms = pickle.load(tf)7. 意味のないPathwayを除去
Pathwayは一連の反応を意味するものなので、例えば1つのタンパクのみ含まれるようなPathwayは下流の解析から除いておくべきです。
同様に、大量のタンパクを含むようなPathway(例えば「細胞内」のような名称のPathway)も除いておくべきであると考えられます。
前節までのようにして取得したSTRINGのPathwayのリストが含むタンパクの数の分布は以下のように可視化できます。
import matplotlib.pyplot as plt pathway_dist = [len(ptm_syms[pt]) for pt in ptm_syms.keys()] plt.hist(pathway_dist, bins=20, log=True) plt.show()
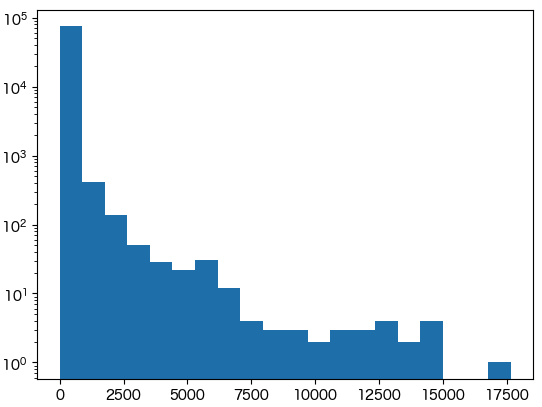
可視化によって、極端な数のタンパクを含むPathwayが少なからず存在することがわかります。
これらは例えば以下のようにして除外しておきます。
# exclude pathways with proteins less than 3 (we do not regard pathways with 1 or 2 proteins as valid pathway.)
pathways_rev1 = [pt for pt in ptm_syms.keys() if 3 <= len(ptm_syms[pt])] # <= 7]
# n_pathway
print(f'n_pathway: {len(pathways)}')
print(f'n_pathway_rev1: {len(pathways_rev1)}')n_pathway: 69736
n_pathway_rev1: 451828. まとめ
本記事では、Pathwayの一覧を取得し、また、各々のPathwayをKeyとして該当Pathwayに含まれるタンパクのシンボル表記のリストをValueに持つ辞書を作成して保存しました。

コメント